炎症性肠病(IBD)包括克罗恩病(CD)和溃疡性结肠炎(UC),肠道纤维化作为其常见且严重的并发症,长期以来被视为慢性炎症的必然归宿。然而,随着对纤维化分子机制认识的不断深化,这一传统观念正在被逐步修正。
当炎症消退,
纤维化为何仍在进展?
在克罗恩病患者中,高达30%的病例最终发展为纤维性狭窄,其中相当比例需要接受手术治疗,而溃疡性结肠炎虽然传统上被认为主要累及黏膜层,但近年研究证实其同样存在显著的黏膜下层纤维化,并与疾病慢性化程度密切相关。值得关注的是,尽管过去十年间生物制剂和小分子药物在控制肠道炎症方面取得了长足进步,但肠道狭窄的发生率并未出现明显下降,这一事实深刻提示:单纯控制炎症并不足以阻断纤维化的进程。
从临床表现来看,肠道纤维化早期往往缺乏特异性症状,直至管腔狭窄引发梗阻性表现时才被察觉,此时组织学改变通常已相当严重。传统观念将纤维化视为慢性炎症的必然归宿,认为瘢痕组织一旦形成便不可逆转,这种认知在很大程度上限制了临床干预的主动性。然而,随着对纤维化分子机制的深入解析,这一“不可逆宿命论”正在被逐步修正,实验证据表明,在去除致纤维化刺激后,早期乃至部分晚期纤维化改变可以发生逆转,这为开发针对性治疗策略提供了理论依据。
炎症独立机制
尽管炎症是纤维化的始动因素,但临床观察与实验研究均提示,炎症与纤维化之间存在“解离”现象。在炎症得到有效控制后,促纤维化因子的水平仍持续维持高位,纤维化进程并不随炎症消退而停止(图1)。这一发现对现有治疗策略提出了根本性挑战:如果纤维化可以在无炎症状态下自我维持和进展,那么单纯抗炎治疗将不足以预防或逆转纤维化。
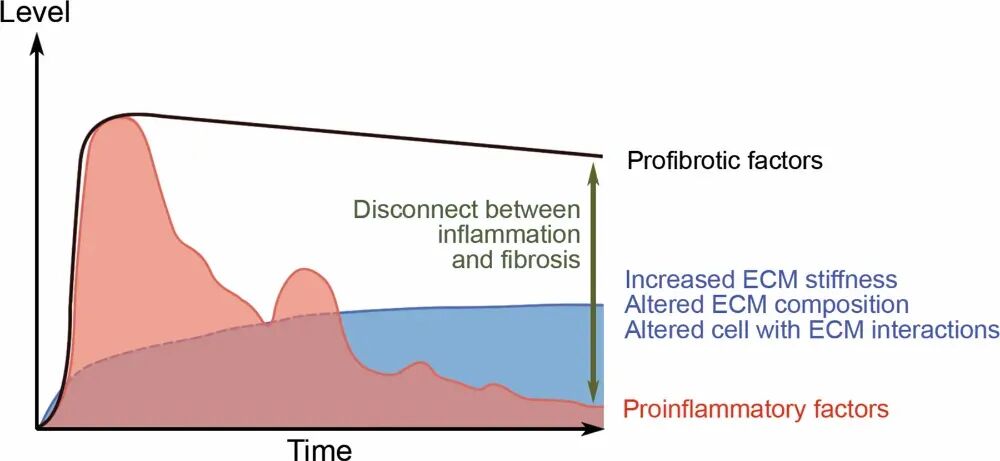
图1炎症消退后,纤维化仍持续进展
CM的机械特性与组成改变是炎症独立纤维化的关键机制。透明质酸在炎症状态下由大分子聚合物降解为低分子量片段,这些片段可结合并激活TLR2和TLR4,触发固有免疫应答,从而在没有持续炎症的情况下维持纤维化信号。ECM作为细胞因子和生长因子的强结合伙伴,可锚定、储存并在适当时机释放TGF-β1等促纤维化因子,形成“因子储备库”。随着狭窄严重程度增加,组织硬度本身即可激活间充质细胞向促纤维化表型转化,间充质细胞的异常收缩又可进一步放大组织硬度,形成自我强化的恶性循环(引用文件2中的图1)。
同型成纤维细胞相互作用为干预提供了新靶点。钙黏蛋白11(CDH11)作为成纤维细胞间的黏附分子,在克罗恩病狭窄部位广泛表达并上调,是CXCL14阳性和MMP/WNT5A阳性成纤维细胞共享的唯一细胞表面受体。体外功能获得与缺失实验、蛋白质组学分析以及基因敲除和抗体介导的阻断实验均证实,CDH11是狭窄性克罗恩病的关键驱动因子和潜在治疗靶点,其抑制可在多种动物模型中减轻纤维化。
间充质细胞衰老是另一个新兴领域。衰老细胞表现出凋亡抵抗和炎性细胞因子、生长因子、免疫调节因子及蛋白酶的分泌增加,可持续维持炎症和纤维化。在复杂型回结肠克罗恩病中,RNA测序数据提示存在与凋亡抵抗和衰老相关分泌表型一致的基因表达谱,PDGFB可在体外诱导成纤维细胞凋亡抵抗。尽管靶向衰老细胞的策略(如senolytic化合物)在皮肤纤维化中显示出前景,但间充质细胞衰老在肠道纤维化中的确切功能表征和贡献仍有待深入探索。
被忽视的纤维化“帮凶”
肠道微生物群通过模式识别受体(PRRs)激活直接参与纤维化进程。肠道共生菌表达的病原体相关分子模式(PAMPs)包括细菌细胞壁成分、脂蛋白、细菌DNA等,这些分子与广泛表达于免疫和非免疫细胞(包括成熟间充质细胞)的Toll样受体(TLRs)或NOD样受体(NLRs)结合。人肠道原代肌成纤维细胞表达多种PRRs,包括TLR 1-9以及NOD1和NOD2。TLR4配体脂多糖(LPS)可促进肠道成纤维细胞的促纤维化活化,增强NF-κB启动子活性并增加胶原产生,TLR4缺陷小鼠表现出减轻的肠道纤维化。鞭毛蛋白通过TLR5和NLRC4依赖方式促进肠道纤维化,携带NOD2突变的克罗恩病患者倾向于表现狭窄表型。
爬行脂肪(creeping fat)是克罗恩病特有的病理现象,指肠系膜脂肪组织包绕病变肠段,其范围与透壁炎症程度密切相关。切除广泛肠系膜(而非最小范围切除)可显著降低克罗恩病复发率和再手术需求,提示爬行脂肪在狭窄形成中具有重要病理生理作用。克罗恩病爬行脂肪与肠系膜脂肪具有独特的微生物组特征,且该特征具有功能相关性。与对照组相比,克罗恩病爬行脂肪中T和B记忆细胞数量增加,脂肪组织来源的趋化因子可主动招募淋巴细胞。尤为关键的是,爬行脂肪与固有肌层增生密切相关,后者现被认为是管腔狭窄形成的主要因素(图2)。爬行脂肪来源的游离脂肪酸(而非脂肪因子)可选择性诱导人肠道平滑肌细胞增殖,形成爬行脂肪与肠道狭窄之间的正反馈环路。
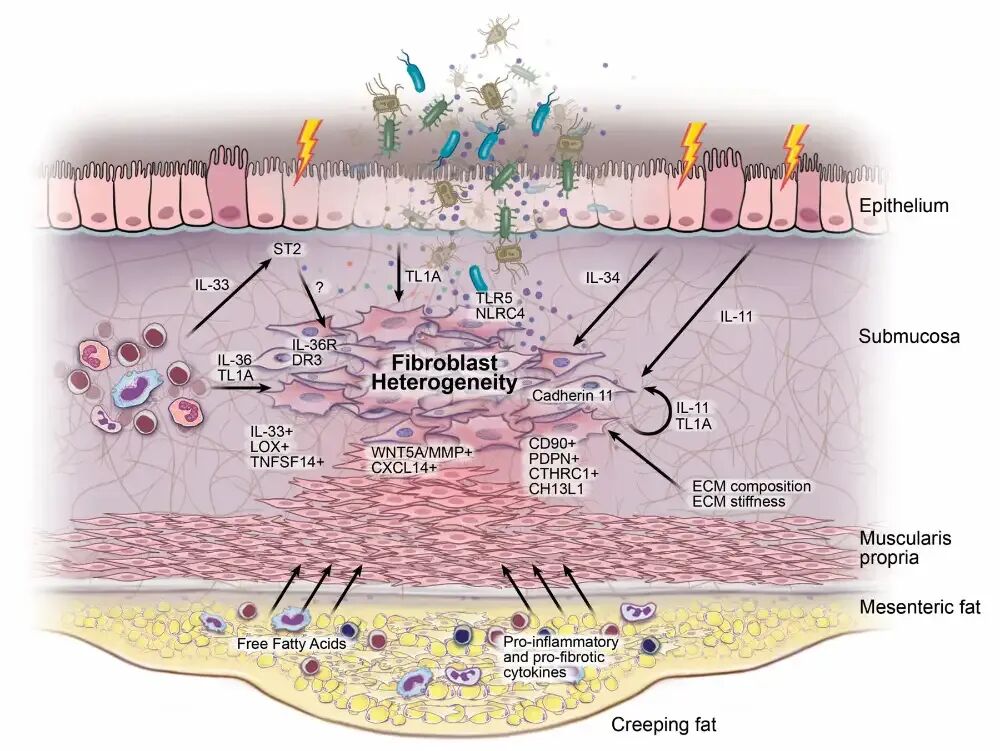
图2肠道纤维化新近机制示意图
溃疡性结肠炎中的纤维化
直至近年,纤维化在溃疡性结肠炎中的发生机制和临床意义仍被严重忽视。传统观点认为溃疡性结肠炎是局限于黏膜/黏膜下层的炎症过程,但一项对706例溃疡性结肠炎组织切片的系统分析显示,所有标本均检测到黏膜下层纤维化,且黏膜下层纤维化与固有肌层增厚与严重和慢性损伤相关,而非与活动性炎症相关,提示纤维化和固有肌层增厚是溃疡性结肠炎进展的常见并发症。即使在内镜下表现正常的溃疡性结肠炎黏膜中,仍存在持续的异常改变,包括ECM重塑、促纤维化细胞因子产生和活化的TGF-β信号通路,这表明尽管炎症是启动纤维化的必要条件,但炎症的抑制和后续愈合并不能阻止纤维化的发展。
治疗困境与突破
目前尚无获批的专门用于预防或逆转IBD相关肠道纤维化的药物,现有管理策略主要依赖控制炎症,但效果有限且短暂,最终往往导致内镜干预或手术切除。其他器官(如肝脏、肺脏)的抗纤维化药物研发已取得显著进展,但肠道纤维化的药物转化明显滞后,主要原因在于:涉及纤维化的细胞类型和信号通路众多,但多数尚未在人体系统中得到验证;缺乏统一的临床试验终点定义;无法准确量化狭窄中的纤维化程度。
TGF-β1通路因其多效性功能而难以直接靶向,直接抑制可能抑制免疫调节、加重炎症并增加恶性肿瘤风险,因此需要探索上下游调控因子的阻断或局部给药策略,如局部递送ALK5抑制剂已进入严格型克罗恩病的临床开发阶段(NCT05843578)。IL-36R抑制剂spesolimab虽在严格性克罗恩病中的临床开发程序因故停止,但其从临床前到临床的探索为后续研究提供了宝贵经验。ROCK抑制剂RXC008作为胃肠道靶向药物,通过干预炎症独立机制在实验性肠道纤维化中显示出前景。
针对特定细胞因子的新型干预策略正在涌现。TL1A阻断抗体在Ⅱ期临床试验中显示出对克罗恩病和溃疡性结肠炎的积极疗效,且临床前研究证实其可减少纤维化。抗IL-17治疗在实验模型中有效但在人体中失败,凸显了动物模型与临床转化的复杂性。Obefazimod在2026年ECCO会议上首次展示了抗纤维化活性证据:在体外人成纤维细胞模型中,Pro-C3(活性纤维化标志物)降低约50%,αSMA(成纤维细胞活化标志物)降低约30%;在体内动物模型中,早期和晚期治疗分别使胶原沉积降低约55%和45%,纤维化评分降低约90%和60%。
靶向ECM组分的新策略开辟了独特路径。山东大学团队发现纤连蛋白是所有实验条件下纤维化肠组织相较正常组织唯一持续高表达的ECM组分,是纤维化ECM的核心结构支架。特异性敲除成纤维细胞中的Fn1基因可将难治性纤维化增厚转化为可逆性炎症性增厚,阻断纤连蛋白可直接抑制其基质组装过程并破坏后续胶原纤维形成,减弱α5β1整合素介导的机械转导,从而打破肠纤维化的自我强化循环。
多组学整合策略为精准药物开发提供了路线图。通过转录组、基因组、表观基因组、蛋白质组、细胞谱系和空间转录组学的多维度整合,可精确定位纤维化相关细胞候选者和通路,构建高分辨率的肠道“组学”图谱,并与其他纤维化疾病的数据集进行比对,从而指导理性药物开发(引用文件2中的图5)。
结语
炎症性肠病肠道纤维化的研究正在经历从“不可逆瘢痕”到“可干预病理过程”的认知转变。这一转变建立在多重机制解析的基础之上:我们认识到肌成纤维细胞来源的多元性,理解了炎症与纤维化解离的病理生理意义,发现了微生物和脂肪组织的促纤维化作用,并初步验证了多个可成药靶点。然而,从机制解析到临床转化的道路依然漫长,动物模型与人类疾病的差异、单一靶点与多因子网络的矛盾、炎症控制与纤维化逆转的分离,均是必须跨越的障碍。
参考文献
[1]RIEDER F, MUKHERJEE PK, MASSEY WJ,et al. Fibrosis in IBD: from pathogenesis to therapeutic targets[J]. Gut,2024;73(5):854-866.DOI:10.1136/gutjnl-2023-329963.
[2]RIEDER F, FIOCCHI C.Intestinal fibrosis in IBD--a dynamic, multifactorial process[J]. Nat Rev Gastroenterol Hepatol,2009;6(4):228-35.DOI:10.1038/nrgastro.2009.31.
ldquo;医学论坛网”发布医学领域研究成果和解读,供专业人员科研参考,不作为诊疗标准,使用需根据具体情况评估。
作者提示:健康医疗分享,仅供参考
(文章中部分图片由AI生成)
炎症性肠病(IBD)包括克罗恩病(CD)和溃疡性结肠炎(UC),肠道纤维化作为其常见且严重的并发症,长期以来被视为慢性炎症的必然归宿。然而,随着对纤维化分子机制认识的不断深化,这一传统观念正在被逐步修正。
当炎症消退,
纤维化为何仍在进展?
在克罗恩病患者中,高达30%的病例最终发展为纤维性狭窄,其中相当比例需要接受手术治疗,而溃疡性结肠炎虽然传统上被认为主要累及黏膜层,但近年研究证实其同样存在显著的黏膜下层纤维化,并与疾病慢性化程度密切相关。值得关注的是,尽管过去十年间生物制剂和小分子药物在控制肠道炎症方面取得了长足进步,但肠道狭窄的发生率并未出现明显下降,这一事实深刻提示:单纯控制炎症并不足以阻断纤维化的进程。
从临床表现来看,肠道纤维化早期往往缺乏特异性症状,直至管腔狭窄引发梗阻性表现时才被察觉,此时组织学改变通常已相当严重。传统观念将纤维化视为慢性炎症的必然归宿,认为瘢痕组织一旦形成便不可逆转,这种认知在很大程度上限制了临床干预的主动性。然而,随着对纤维化分子机制的深入解析,这一“不可逆宿命论”正在被逐步修正,实验证据表明,在去除致纤维化刺激后,早期乃至部分晚期纤维化改变可以发生逆转,这为开发针对性治疗策略提供了理论依据。
炎症独立机制
尽管炎症是纤维化的始动因素,但临床观察与实验研究均提示,炎症与纤维化之间存在“解离”现象。在炎症得到有效控制后,促纤维化因子的水平仍持续维持高位,纤维化进程并不随炎症消退而停止(图1)。这一发现对现有治疗策略提出了根本性挑战:如果纤维化可以在无炎症状态下自我维持和进展,那么单纯抗炎治疗将不足以预防或逆转纤维化。
图1炎症消退后,纤维化仍持续进展
CM的机械特性与组成改变是炎症独立纤维化的关键机制。透明质酸在炎症状态下由大分子聚合物降解为低分子量片段,这些片段可结合并激活TLR2和TLR4,触发固有免疫应答,从而在没有持续炎症的情况下维持纤维化信号。ECM作为细胞因子和生长因子的强结合伙伴,可锚定、储存并在适当时机释放TGF-β1等促纤维化因子,形成“因子储备库”。随着狭窄严重程度增加,组织硬度本身即可激活间充质细胞向促纤维化表型转化,间充质细胞的异常收缩又可进一步放大组织硬度,形成自我强化的恶性循环(引用文件2中的图1)。
同型成纤维细胞相互作用为干预提供了新靶点。钙黏蛋白11(CDH11)作为成纤维细胞间的黏附分子,在克罗恩病狭窄部位广泛表达并上调,是CXCL14阳性和MMP/WNT5A阳性成纤维细胞共享的唯一细胞表面受体。体外功能获得与缺失实验、蛋白质组学分析以及基因敲除和抗体介导的阻断实验均证实,CDH11是狭窄性克罗恩病的关键驱动因子和潜在治疗靶点,其抑制可在多种动物模型中减轻纤维化。
间充质细胞衰老是另一个新兴领域。衰老细胞表现出凋亡抵抗和炎性细胞因子、生长因子、免疫调节因子及蛋白酶的分泌增加,可持续维持炎症和纤维化。在复杂型回结肠克罗恩病中,RNA测序数据提示存在与凋亡抵抗和衰老相关分泌表型一致的基因表达谱,PDGFB可在体外诱导成纤维细胞凋亡抵抗。尽管靶向衰老细胞的策略(如senolytic化合物)在皮肤纤维化中显示出前景,但间充质细胞衰老在肠道纤维化中的确切功能表征和贡献仍有待深入探索。
被忽视的纤维化“帮凶”
肠道微生物群通过模式识别受体(PRRs)激活直接参与纤维化进程。肠道共生菌表达的病原体相关分子模式(PAMPs)包括细菌细胞壁成分、脂蛋白、细菌DNA等,这些分子与广泛表达于免疫和非免疫细胞(包括成熟间充质细胞)的Toll样受体(TLRs)或NOD样受体(NLRs)结合。人肠道原代肌成纤维细胞表达多种PRRs,包括TLR 1-9以及NOD1和NOD2。TLR4配体脂多糖(LPS)可促进肠道成纤维细胞的促纤维化活化,增强NF-κB启动子活性并增加胶原产生,TLR4缺陷小鼠表现出减轻的肠道纤维化。鞭毛蛋白通过TLR5和NLRC4依赖方式促进肠道纤维化,携带NOD2突变的克罗恩病患者倾向于表现狭窄表型。
爬行脂肪(creeping fat)是克罗恩病特有的病理现象,指肠系膜脂肪组织包绕病变肠段,其范围与透壁炎症程度密切相关。切除广泛肠系膜(而非最小范围切除)可显著降低克罗恩病复发率和再手术需求,提示爬行脂肪在狭窄形成中具有重要病理生理作用。克罗恩病爬行脂肪与肠系膜脂肪具有独特的微生物组特征,且该特征具有功能相关性。与对照组相比,克罗恩病爬行脂肪中T和B记忆细胞数量增加,脂肪组织来源的趋化因子可主动招募淋巴细胞。尤为关键的是,爬行脂肪与固有肌层增生密切相关,后者现被认为是管腔狭窄形成的主要因素(图2)。爬行脂肪来源的游离脂肪酸(而非脂肪因子)可选择性诱导人肠道平滑肌细胞增殖,形成爬行脂肪与肠道狭窄之间的正反馈环路。
图2肠道纤维化新近机制示意图
溃疡性结肠炎中的纤维化
直至近年,纤维化在溃疡性结肠炎中的发生机制和临床意义仍被严重忽视。传统观点认为溃疡性结肠炎是局限于黏膜/黏膜下层的炎症过程,但一项对706例溃疡性结肠炎组织切片的系统分析显示,所有标本均检测到黏膜下层纤维化,且黏膜下层纤维化与固有肌层增厚与严重和慢性损伤相关,而非与活动性炎症相关,提示纤维化和固有肌层增厚是溃疡性结肠炎进展的常见并发症。即使在内镜下表现正常的溃疡性结肠炎黏膜中,仍存在持续的异常改变,包括ECM重塑、促纤维化细胞因子产生和活化的TGF-β信号通路,这表明尽管炎症是启动纤维化的必要条件,但炎症的抑制和后续愈合并不能阻止纤维化的发展。
治疗困境与突破
目前尚无获批的专门用于预防或逆转IBD相关肠道纤维化的药物,现有管理策略主要依赖控制炎症,但效果有限且短暂,最终往往导致内镜干预或手术切除。其他器官(如肝脏、肺脏)的抗纤维化药物研发已取得显著进展,但肠道纤维化的药物转化明显滞后,主要原因在于:涉及纤维化的细胞类型和信号通路众多,但多数尚未在人体系统中得到验证;缺乏统一的临床试验终点定义;无法准确量化狭窄中的纤维化程度。
TGF-β1通路因其多效性功能而难以直接靶向,直接抑制可能抑制免疫调节、加重炎症并增加恶性肿瘤风险,因此需要探索上下游调控因子的阻断或局部给药策略,如局部递送ALK5抑制剂已进入严格型克罗恩病的临床开发阶段(NCT05843578)。IL-36R抑制剂spesolimab虽在严格性克罗恩病中的临床开发程序因故停止,但其从临床前到临床的探索为后续研究提供了宝贵经验。ROCK抑制剂RXC008作为胃肠道靶向药物,通过干预炎症独立机制在实验性肠道纤维化中显示出前景。
针对特定细胞因子的新型干预策略正在涌现。TL1A阻断抗体在Ⅱ期临床试验中显示出对克罗恩病和溃疡性结肠炎的积极疗效,且临床前研究证实其可减少纤维化。抗IL-17治疗在实验模型中有效但在人体中失败,凸显了动物模型与临床转化的复杂性。Obefazimod在2026年ECCO会议上首次展示了抗纤维化活性证据:在体外人成纤维细胞模型中,Pro-C3(活性纤维化标志物)降低约50%,αSMA(成纤维细胞活化标志物)降低约30%;在体内动物模型中,早期和晚期治疗分别使胶原沉积降低约55%和45%,纤维化评分降低约90%和60%。
靶向ECM组分的新策略开辟了独特路径。山东大学团队发现纤连蛋白是所有实验条件下纤维化肠组织相较正常组织唯一持续高表达的ECM组分,是纤维化ECM的核心结构支架。特异性敲除成纤维细胞中的Fn1基因可将难治性纤维化增厚转化为可逆性炎症性增厚,阻断纤连蛋白可直接抑制其基质组装过程并破坏后续胶原纤维形成,减弱α5β1整合素介导的机械转导,从而打破肠纤维化的自我强化循环。
多组学整合策略为精准药物开发提供了路线图。通过转录组、基因组、表观基因组、蛋白质组、细胞谱系和空间转录组学的多维度整合,可精确定位纤维化相关细胞候选者和通路,构建高分辨率的肠道“组学”图谱,并与其他纤维化疾病的数据集进行比对,从而指导理性药物开发(引用文件2中的图5)。
结语
炎症性肠病肠道纤维化的研究正在经历从“不可逆瘢痕”到“可干预病理过程”的认知转变。这一转变建立在多重机制解析的基础之上:我们认识到肌成纤维细胞来源的多元性,理解了炎症与纤维化解离的病理生理意义,发现了微生物和脂肪组织的促纤维化作用,并初步验证了多个可成药靶点。然而,从机制解析到临床转化的道路依然漫长,动物模型与人类疾病的差异、单一靶点与多因子网络的矛盾、炎症控制与纤维化逆转的分离,均是必须跨越的障碍。
参考文献
[1]RIEDER F, MUKHERJEE PK, MASSEY WJ,et al. Fibrosis in IBD: from pathogenesis to therapeutic targets[J]. Gut,2024;73(5):854-866.DOI:10.1136/gutjnl-2023-329963.
[2]RIEDER F, FIOCCHI C.Intestinal fibrosis in IBD--a dynamic, multifactorial process[J]. Nat Rev Gastroenterol Hepatol,2009;6(4):228-35.DOI:10.1038/nrgastro.2009.31.
ldquo;医学论坛网”发布医学领域研究成果和解读,供专业人员科研参考,不作为诊疗标准,使用需根据具体情况评估。
作者提示:健康医疗分享,仅供参考
(文章中部分图片由AI生成)
